Noticia Notiweb (Madri+D): Identificado un mecanismo molecular implicado en la degeneración neuronal provocada por la enfermedad de Huntington
Estos resultados podrían servir para diseñar nuevas terapias para tratar esta y otras enfermedades que afectan al cerebro
Investigadores del Instituto de Neurociencias de la Universidad de Barcelona (UBNeuro) y el Instituto de Investigaciones Biomédicas August Pi i Sunyer (IDIBAPS) (Catalunya, España) han descrito un mecanismo, el aumento de síntesis proteica, que participa en la degeneración del tipo de neuronas afectadas en la enfermedad de Huntington, una patología genética neurodegenerativa. Estos resultados, publicados en la revista científica Brain, podrían servir para diseñar nuevas terapias para tratar esta y otras enfermedades que afectan al cerebro.
El trabajo está liderado por Esther Pérez Navarro, profesora de la Facultad de Medicina y Ciencias de la Salud de la UB e investigadora IDIBAPS. También han participado en el estudio investigadores de la Universidad Pablo de Olavide.
La enfermedad de Huntington es un trastorno neurodegenerativo genético causado por la mutación del gen de la huntingtina, que provoca la pérdida precoz de las neuronas estriatales de proyección, con efectos en la coordinación motora y deterioro cognitivo y psiquiátrico. El nuevo trabajo ha analizado el rol que en este proceso tiene la alteración de la síntesis proteica, un mecanismo que permite a las neuronas leer el código genético para sintetizar proteínas.
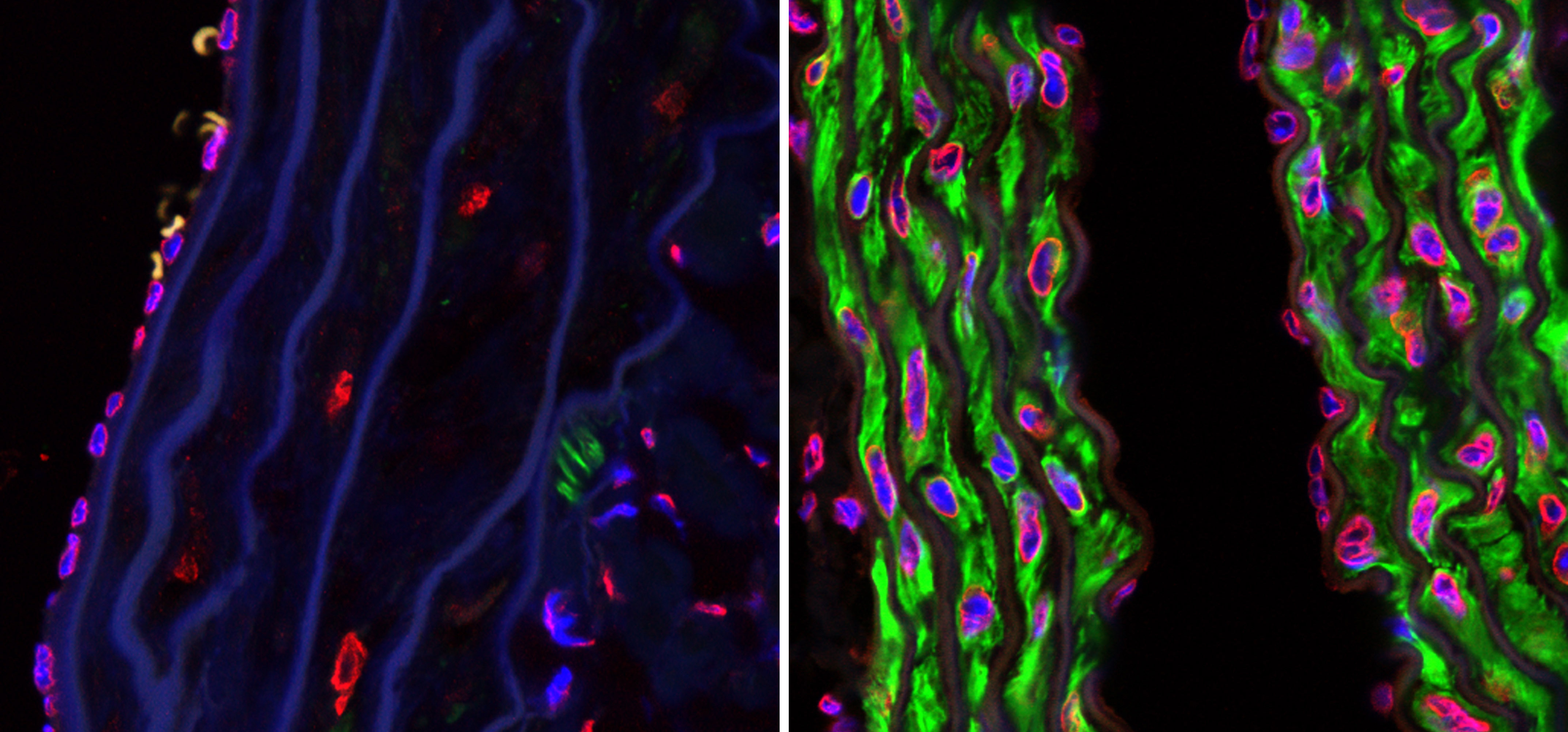
Para estudiar este mecanismo se analizaron los niveles totales y fosforilados de 4E-BP1, una proteína que inhibe la síntesis proteica, en un modelo de ratón de la enfermedad. «Los resultados muestran que los niveles totales de esta proteína se reducen, mientras que los niveles de fosforilación aumentan, en las neuronas estriatales de proyección de los ratones con la dolencia, en comparación con los ratones control, por lo que aumenta la síntesis de proteínas, cosa que igualmente encontramos en muestras de cerebros de pacientes», explica Esther Pérez Navarro, también investigadora del Centro de Investigación Biomédica en Red sobre Enfermedades Neurodegenerativas (CIBERNED).
Para confirmar esta relación entre la actividad inapropiada de la síntesis proteica y la enfermedad, los investigadores bloquearon farmacológicamente este mecanismo y comprobaron que mejoraba la función motora de los ratones y que se restablecían los niveles normales de diferentes valores moleculares en el cerebro. «Estos resultados indican que un aumento de la síntesis proteica en la enfermedad de Huntington es perjudicial y, por tanto, supone una potencial diana terapéutica para nuevos tratamientos, como por ejemplo un fármaco que pueda suministrarse de forma no invasiva para normalizar la síntesis proteica», detalla la investigadora.
Aunque es la primera vez que se relaciona la alteración de la síntesis proteica con la enfermedad de Huntington, se trata de un mecanismo que se ha descrito en otras enfermedades neurodegenerativas (como el alzhéimer y el párkinson) y en otros trastornos mentales, como el autismo. «El hecho de encontrar mecanismos comunes a distintas patologías que afectan a nuestro cerebro hace más atractivo el hallazgo, ya que la misma terapia podría ser beneficiosa para varias enfermedades», destaca la investigadora.
Esta investigación también abre la puerta a la identificación de biomarcadores que permitan detectar la enfermedad antes de que aparezcan los primeros síntomas. En este sentido, los investigadores, en colaboración con la Unidad de Párkinson y Trastornos del Movimiento del Hospital de la Santa Creu i Sant Pau, están estudiando si la síntesis proteica también está alterada en células fuera del cerebro, como las sanguíneas y los fibroblastos (células de la piel). «La ventaja de realizar este estudio con una enfermedad como la de Huntington, que está asociada a una mutación genética, es que podemos analizar estos cambios en individuos portadores que aún no presentan síntomas y hacer un seguimiento a lo largo del tiempo», concluye la investigadora.
FUENTE: NOTIWEB.