Buenas noticias de un proyecto financiado por Fundación Isabel Gemio, Federación ASEM y FEDER – Noticia Ciber ISCIII
Hallan una terapia para una rara enfermedad cardíaca: la miocardiopatía arritmogénica tipo 5
La miocardiopatía arritmogénica tipo 5 es una enfermedad genética letal para la que desgraciadamente no existe cura. Ahora, investigadores del CIBERCV, del CNIC y del Hospital Puerta de Hierro Majadahonda (Madrid) han encontrado un posible tratamiento para esta enfermedad rara. Los investigadores, cuyo trabajo se publica en Circulation, han visto en un modelo de ratón que inhibir una proteína, la quinasa GSK3b, reduce la fibrosis y mejora la función cardiaca.
La miocardiopatía arritmogénica puede producir muerte súbita, sobre todo en hombres jóvenes. Tanto los hombres que no sufren muerte súbita como las mujeres que padecen esta enfermedad desarrollan con el tiempo insuficiencia cardiaca, explican los coordinadores del estudio, Enrique Lara Pezzi, jefe de grupo en el CNIC, y Pablo García-Pavía, jefe de grupo del CIBERCV y director de la Unidad de Cardiopatías Familiares del Servicio de Cardiología del Hospital Universitario Puerta de Hierro.
Se calcula que la miocardiopatía arritmogénica la padece entre el 0,02% y el 0,1% en la población general, por lo que se considera una enfermedad rara. El subtipo más agresivo de esta enfermedad se denomina miocardiopatía arritmogénica tipo 5 y se debe a una alteración genética en el gen TMEM43. Aunque los primeros pacientes con miocardiopatía arritmogénica tipo 5 se identificaron en la isla de Terranova (Canadá), se ha detectado también en otras zonas del mundo, incluida España.
- Se calcula que la miocardiopatía arritmogénica la padece entre el 0,02% y el 0,1% en la población general, por lo que se considera una enfermedad rara
Durante las etapas iniciales, ‘la fase oculta’, señalan los investigadores españoles, los pacientes no suelen tener síntomas, aunque ya presentan riesgo de padecer arritmias y sufrir una muerte súbita. A pesar de que el ventrículo derecho es el más afectado en fases iniciales, a medida que se expande la fibrosis puede comprometer también el ventrículo izquierdo y aparecen síntomas y manifestaciones de insuficiencia cardiaca que hacen que los pacientes puedan requerir un trasplante de corazón.
“Sin embargo, no se conocen los mecanismos por los que se produce esta enfermedad y, a día de hoy, no existe cura”, apunta el Dr. Lara Pezzi. Ello hace que el tratamiento sea fundamentalmente paliativo y se base en la prevención de la muerte súbita mediante la implantación de un desfibrilador automático implantable (DAI) y, posteriormente, en el manejo de la insuficiencia cardiaca, incluyendo el trasplante cardiaco.
En un claro ejemplo de investigación traslacional, los grupos del Dr. Lara Pezzi y del Dr. García-Pavía estudiaron esta enfermedad con el fin de hallar nuevos tratamientos que pudiesen ser aplicados a los pacientes con esta devastadora enfermedad y que habían sido diagnosticados por primera vez en España en el hospital Puerta de Hierro. “Nos habíamos encontrado una enfermedad de la que se sabía muy poco y en la que múltiples personas de la misma familia fallecían muy jóvenes” declara el Dr García-Pavía.
“Nos dimos cuenta que necesitábamos entender mejor la enfermedad desde el principio para poder buscar tratamientos eficaces y, para eso, necesitábamos disponer de un animal que padeciese la enfermedad y que pudiésemos estudiar desde su nacimiento” prosigue el Dr García-Pavía. Así, fruto de esta necesidad clínica los dos grupos de investigación decidieron colaborar para desarrollar un modelo transgénico de ratón que expresase la proteína humana TMEM43. Juntos lograron crear animales que desarrollan la enfermedad humana. De esta manera, encontraron que la versión mutante de TMEM43 provoca la activación de una proteína, la quinasa GSK3b, que causa la muerte progresiva de las células cardiacas, que son sustituidas poco a poco por fibrosis, una de los rasgos más característicos de esta enfermedad. “Al cabo de pocos meses, el corazón no tiene suficientes células que funcionen de forma adecuada y bombeen la sangre eficazmente, por lo que el animal muere a causa de una insuficiencia cardiaca”, explica Laura Padrón-Barthe, primera autora del artículo.
- Los investigadores están utilizando su modelo de ratón para evaluar la eficacia de medicamentos que se usan en humanos que padecen insuficiencia cardiaca
En la búsqueda de una terapia, los investigadores testaron diversos tratamientos en el modelo de ratón. Mientras que el tratamiento de la fibrosis como tal no dio resultados positivos, la inhibición de GSK3b mediante dos estrategias distintas -un inhibidor farmacológico o la sobrexpresión de una subunidad de la calcineurina CnAβ1- sí obtuvo resultados. “Ambas aproximaciones redujeron la muerte de las células cardiacas, mejoraron la contracción del corazón y prolongaron la supervivencia de los ratones”, comentan los autores.
No obstante, los investigadores advierten que, aunque este modelo de ratón transgénico es el único que reproduce ARVC5 humano, no presenta ciertas características de la enfermedad humana ya que, por ejemplo, no se encontraron diferencias significativas entre machos y hembras, en contraste con los pacientes humanos en los que la enfermedad es mucho más agresiva entre varones.
En cualquier caso, una vez conocida una posible vía eficaz para tratar la enfermedad en los ratones, los investigadores están trabajando nuevamente juntos para trasladar sus hallazgos a los pacientes. Así, están utilizando este modelo de ratón para evaluar la eficacia de medicamentos que se usan en humanos que padecen insuficiencia cardiaca, con el fin de averiguar si serían útiles para tratar la miocardiopatía arritmogénica tipo 5. Además, están analizando estrategias de terapia génica que puedan mejorar la función cardiaca e incluso curar la enfermedad.
El estudio ha sido financiado por proyectos del Ministerio de Ciencia, Innovación y Universidades, el Instituto de Salud Carlos III, la Comunidad de Madrid, la Sociedad Española de Cardiología y la Fundación Isabel Gemio “Todos somos raros”.
Artículo de referencia
Laura Padrón-Barthe, María Villalba-Orero, Jesús M. Gómez-Salinero, Fernando Domínguez, Marta Román, Javier Larrasa-Alonso, Paula Ortiz-Sánchez, Fernando Martínez, Marina López-Olañeta, Elena Bonzón-Kulichenko, Jesús Vázquez, Carlos Martí-Gómez, Demetrio J. Santiago, Belén Prados, Giovanna Giovinazzo, María Victoria Gómez-Gaviro, Silvia Priori, … Severe Cardiac Dysfunction and Death Caused by ARVC Type 5 is Improved by Inhibition of GSK3β https://doi.org/10.1161/CIRCULATIONAHA.119.040366 Circulation
*Imagen:
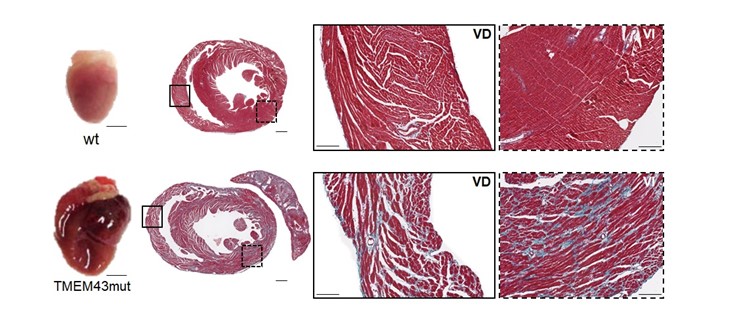
La expresión de la proteína TMEM43 humana con la mutación p.S358L en el corazón del ratón causa fibrosis cardiaca y dilatación del corazón. Fila superior, corazones de ratones control (wt, “wild type”). Fila inferior, corazones de ratones que expresan la proteína TMEM43 mutante (TMEM43mut)

De izda. a dcha Marta Román, Fernando Domínguez, Laura Padrón-Barthe, Pablo García-Pavía, Giovanna Giovinazzo, Enrique Lara-Pezzi, Demetrio J. Santiago y Javier Larrasa-Alonso. – CNIC


