Noticias sobre un nuevo estudio que pertenece al grupo de investigación que también se encuentra entre nuestras líneas de investigación financiada: BASES MOLECULARES DE ENFERMEDADES NEUROMETABÓLICAS Y DESARROLLO DE TERAPIAS ESPECÍFICAS DE MUTACIÓN. El proyecto que recibe nuestra ayuda se apuesta por la investigación con las nuevas técnicas de secuenciación masiva del genoma para acelerar el proceso de diagnóstico de estas patologías en estudio. A la par, se avanzará en el desarrollo de terapias con dos tipos de fármacos: las chaperonas farmacológicas y la terapia antisentido. Ambas terapias están dirigidas a corregir defectos genéticos concretos y podrían aplicarse a numerosas enfermedades genéticas ya que todas ellas comparten mecanismos de acción. Estos avances contribuirán a incrementar el interés de las compañías farmacéuticas en el desarrollo de medicamentes catalogados como “huérfanos”.
En esta ocasión el trabajo recogido en esta noticia y publicado en la revista PLoS ONE, realizado por Ainhoa Martínez y dirigido por la Dra. Eva Richard, pertenecientes al grupo de investigación anteriormente mencionado; el objetivo principal ha sido avanzar en el conocimiento de la fisiopatología de la homocistinuria; es decir, en el conocimiento de los mecanismos celulares que pueden estar implicados en la enfermedad, analizando, para ello, diversos parámetros relacionados con el estrés oxidativo del RE, la homeostasis del calcio y la autofagia en fibroblastos de cinco pacientes homocistinúricos con defectos en la vía de remetilación de la homocisteína.
Puedes leer todo el artículo en el siguiente enlace: NUEVAS TERAPIAS CON ANTIOXIDANTES
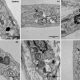 Análisis de la ultraestructura mitocondrial mediante microscopía electrónica. Fibroblastos de un individuo control y cinco pacientes con homocistinuria. En los pacientes se observa (flechas) que las mitocondrias están siendo degradadas en autofagosomas | UAM Gazette
Análisis de la ultraestructura mitocondrial mediante microscopía electrónica. Fibroblastos de un individuo control y cinco pacientes con homocistinuria. En los pacientes se observa (flechas) que las mitocondrias están siendo degradadas en autofagosomas | UAM Gazette