Compartimos con vosotros las publicaciones científicas de la Dra. Isabel Illa Sendra, miembro de nuestro Comité Científico. Illa es también Jefa de la Unidad de Enfermedades Neuromusculares del Servicio Neurología del Hospital Santa Creu i Sant Pau de Barcelona y Catedrática de Neurología de la Universidad Autónoma de Barcelona. Además, dirige el Laboratorio de Neurología Experimental del Institut de Recerca del HSCSP y es miembro de CIBERER.
En el cuerpo de esta noticia ofrecemos un resumen en castellano de algunas de sus publicaciones científicas. Más abajo, tras cada resumen, hay un enlace donde puedes descargártelas.
La reconstitución parcial de disferlina mediante mesoangioblastos murinos adultos es suficiente para la recuperación de la plena funcionalidad en un modelo murino de disferlinopatía
J Díaz-Manera, T Touvier, A Dellavalle, R Tonlorenzi, FS Tedesco, G Messina M Meregalli, C Navarro, L Perani,C Bonfanti, I Illa, Y Torrente, G Cossu
Cell Death and Disease (2010) 1, e61
La deficiencia de disferlina conduce a una forma peculiar de distrofia muscular debida a un defecto en la reparación del sarcolema y en la actualidad carece de una terapia. Hemos desarrollado un protocolo de terapia celular con mesoangioblastos adultos murinos de tipo salvaje. Estas células se diferencian con alta eficiencia en el músculo esquelético in vitro y se distinguen de las células satélite, ya que no expresan Pax7. Después de la administración intramuscular o intraarterial de mesoangioblastos tipo salvaje a ratones SCID / Blaj, un nuevo modelo disferlinopatía, éstos colonizaron de manera eficiente los músculos distróficos y restauraron parcialmente la expresión de disferlina. Sin embargo, los ensayos funcionales realizados en fibras individuales aisladas de los músculos trasplantados mostraron una capacidad de reparación normal de la membrana después de las lesiones inducidas por láser. Este resultado, que refleja la corrección de un déficit enzimático más que estructural, sugiriendo que esta miopatía puede ser más fácil de tratar con terapia celular o génica que otras formas de distrofia muscular.
Descargar publicación sobre la reconstitución parcial de disferlina mediante mesoangioblastos murinos
Comparación de la expresión de disferlina en músculo esquelético humano con la de monocitos para el diagnóstico de la miopatía por déficit de disferlina
E Gallardo, N de Luna, J Diaz-Manera, R Rojas-García, L Gonzalez-
Quereda, B Flix, A de Morrée, S van der Maarel, I Illa
PLoS ONE (2011) 6(12): e29061.
Las disferlinopatías son causados por mutaciones en el gen DYSF. El diagnóstico es complejo debido a la alta variabilidad clínica de la expresión dysferlin enfermedad y porque en la biopsia de músculo la expresión de disferlina puede estar reducida de forma secundaria a un defecto primario en algún otro gen. La disferlina también se expresa en monocitos de sangre periférica (PBM). El estudio de disferlina en monocitos se utiliza para el diagnóstico de las disferlinopatías. El objetivo del estudio fue determinar si la expresión de disferlina en PBM se correlaciona con la de músculo esquelético. Mediante Western-blot (WB) se cuantificó la expresión de disferlina en PBM de 21 pacientes con otras miopatías en los que se excluyeron mutaciones en DYSF y de 17 pacientes que tenían disferlinopatía y dos mutaciones en DYSF. Los resultados se compararon con la expresión de proteínas en el músculo por WB e inmunohistoquímica (IH). Hemos encontrado una buena correlación entre el músculo esquelético y monocitos utilizando WB. Sin embargo, los resultados eran engañosos utilizando IH porque la expresión anormal de disferlina también se observó en 13/21 controles patológicos. El análisis de la expresión de disferlina en PBM es útil cuando: 1) el patrón de IH en músculo esquelético es anormal o 2) cuando el WB de músculo no se puede realizar ya sea porque se carece de muestra o ésta es insuficiente o porque la biopsia de músculo se ha tomado de un músculo en una etapa terminal y por tanto se compone principalmente de grasa y tejido fibrótico.
Descargar publicación sobre comparación de la expresión de disferlina en músculo esquelético
La 1α, 25 (OH) 2-vitamina D3 aumenta la expresión de disferlina in vitro y en un ensayo clínico en humanos
N De Luna, J Díaz-Manera, C Paradas, C Iturriaga, R Rojas-García, J Araque, M Genebriera, I Gich, I Illa and E Gallardo
Molecular Therapy (2012) 20 (10): 1988–1997
Las disferlinopatías son un grupo heterogéneo de distrofias musculares hereditarias autosómicas recesivas causadas por mutaciones en el gen DYSF. La disferlina se expresa principalmente en el músculo esquelético y en los monocitos y los pacientes muestran una grave reducción o ausencia de la proteína en ambos tejidos. La vitamina D3 promueve la diferenciación de células HL60 provenientes de una leucemia promielocítica. Se analizó el efecto de la vitamina D3 en expresión de disferlina in vitro utilizando células HL60, monocitos y miotubos de controles y portadores de una mutación en DYSF. También se realizó un estudio observacional con vitamina D3 oral en una cohorte de 21 portadores. Quince sujetos fueron tratados durante 1 año y la expresión de disferlina en monocitos se analizó antes y después del tratamiento. El tratamiento con vitamina D3 aumentó la expresión de disferlina in vitro. El efecto de la vitamina D3 fue mediada tanto por una vía no genómica a través de MEK / ERK y una vía genómica que implica la unión del receptor de la vitamina D3 al promotor de disferlina. Los portadores tratados con vitamina D3 habían aumentado significativamente la expresión de disferlina en monocitos en comparación con los portadores no tratados (P <0,05). Estos hallazgos tienen importantes implicaciones terapéuticas ya que una combinación de diferentes estrategias moleculares junto con el tratamiento con vitamina D3 podría aumentar la expresión de disferlina hasta niveles de proteína funcionales.
Descargar publicación sobre La 1α, 25 (OH) 2-vitamina D3 aumenta la expresión de disferlina in vitro
La disferlina interacciona con calsecuestrina-1, miomesina-2 y dineína en músculo esquelético humano
B Flix, C de la Torre, J Castillo, C Casal, I Illa, E Gallardo
The International Journal of Biochemistry & Cell Biology 45 (2013) 1927– 1938
Las disferlinopatías son un grupo de distrofias musculares progresivas caracterizadas por mutaciones en el gen DYSF. Estas mutaciones causan la disminución o la ausencia completa de disferlina, una proteína que se expresa en el músculo esquelético y desempeña un papel en la reparación de la membrana. Nuestro objetivo era analizar las proteínas que constituyen un complejo con la disferlina y su interacción dentro del complejo mediante ensayos de inmunoprecipitación (IP), electroforesis en geles Blue Native (BN) en músculo esquelético de adultos sanos y y en cultivos de miotubos control, y análisis fluorescence lifetime imaging–fluorescence resonance energy transfer (Flim-FRET) en miotubos sanos. La combinación de inmunoprecipitación y electroforesis BN nos permitió identificar proteínas que ya se conocía que interaccionaban con la disferlina – tales como la caveolina-3, AHNAK, anexinas, o Trim72 / MG53 – y nuevas proteínas del complejo. Las imágenes de FLIM mostraron una interacción directa de disferlina con Trim72 / MG53, AHNAK, dineína citoplasmática, myomesin-2 y calsecuestrina-1, pero no con la caveolina-3 o distrofina. En conclusión, a pesar de que la IP y BN son herramientas útiles para identificar las proteínas en un complejo, se necesitan técnicas como FLIM-FRET para determinar las interacciones directas e indirectas de estas proteínas dentro del complejo. Este conocimiento nos puede ayudar a comprender mejor el papel de la disferlina en el tejido muscular e identificar nuevos genes implicados en distrofias musculares en la que el gen responsable es todavía desconocido.
Descargar publicacion La disferlina interacciona con calsecuestrina-1, miomesina-2 y dineína en músculo esquelético humano
El transplante de medula ósea en un modelo murino de disferlinopatía produce una mejora funcional leve
B Flix, X Suárez-Calvet, J Díaz-Manera, E Santos-Nogueira, R Mancuso, J Barquinero, M Navas, X Navarro, I Illa, E Gallardo
Stem Cells and Development (2013) 22(21): 2885-2894
Las disferlinopatías son causadas por mutaciones en el gen DYSF. La disferlina es una proteína expresada principalmente en el músculo esquelético y monocitos. La terapia celular constituye una herramienta prometedora para el tratamiento de distrofias musculares. El objetivo de nuestro estudio fue evaluar el efecto del trasplante de médula ósea (TMO) en el modelo de ratón A / J Dysf prmd con disferlinopatía. A tal fin, se estudió la expresión de disferlina por Western Blot y / o inmunohistoquímica en ratones trasplantados y controles. Se realizaron análisis computerizados de locomoción y técnicas electrofisiológicas para probar la mejora funcional. Observamos expresión de disferlina en esplenocitos, pero no en el músculo esquelético de los ratones trasplantados. Sin embargo, la prueba de locomoción, los estudios de electromiografía, y la histología muscular mostraron una mejora en todos los ratones trasplantados que era más significativa en los animales trasplantados con células disferlina + / +. En conclusión, aunque el TMO restaura la expresión disferlina en monocitos, pero no en el músculo esquelético, la función muscular se recuperó parcialmente. Nosotros proponemos que la ligera mejoría observada en los estudios funcionales podría estar relacionado con factores, tales como el factor de crecimiento de hepatocitos, liberado después de trasplante de médula ósea que impidió la degeneración muscular.
Descargar publicación sobre cómo el transplante de medula ósea en un modelo murino de disferlinopatía produce una mejora funcional leve

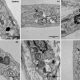 Análisis de la ultraestructura mitocondrial mediante microscopía electrónica. Fibroblastos de un individuo control y cinco pacientes con homocistinuria. En los pacientes se observa (flechas) que las mitocondrias están siendo degradadas en autofagosomas | UAM Gazette
Análisis de la ultraestructura mitocondrial mediante microscopía electrónica. Fibroblastos de un individuo control y cinco pacientes con homocistinuria. En los pacientes se observa (flechas) que las mitocondrias están siendo degradadas en autofagosomas | UAM Gazette